Guillain-Barre syndrome (GBS)
Overview I Aetiology I Clinical features I Complications I Investigations I Treatment I Prognosis
Guillain-barre syndrome (GBS) is a post infectious polyneuropathy that causes demyelination in mainly motor but sometimes sensory nerves also. The syndrome usually develops 1-4 weeks after viral infection and rarely follows surgery and immunization. Guillain-barre syndrome (GBS) is also described as rapid-onset muscle weakness. This is caused by the immune system damaging the peripheral nervous system (PNS). This often spread to the arm upper body, with both sides involved. The cause of Guillain-Baree syndrome (GBS) is unknown. The underlying mechanism involves an autoimmune disorder.
Aetiology
Occurs commonly which gastrointestinal infection (especially Campylobacter jejuni) or respiratory tract infection (mycoplasma pneumoniae)
Clinical features
In the case of acute onset, leg pain and stiffness are common, transiently bladder may be involved.
The characteristics clinical features are symmetrical muscles weakness; weakness begins usually in the lower extremities and progressively involves the trunk, upper limbs, and finally the bulbar muscle (50%), a pattern formerly known as laundry ascending paralysis. The onset is gradual and progresses over days or weeks.
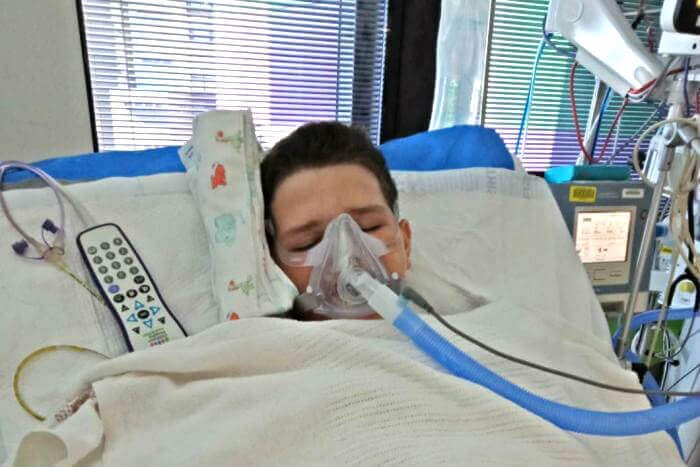
Respiratory insufficiency may result. Dyspnea and facial weakness are impending signs of respiratory failure.
Autonomic manifestations are cardiac arrhythmia, fluctuation of blood pressure and heart rate, postural hypotension, profound bradycardia.
On examination, profound muscle weakness and loss of tendon reflexes occur. The miller-fisher syndrome-consists of acute external ophthalmoplegia, ataxia and areflexia may also be observed.
Complications
- Respiratory paralysis
- Urinary incontinence or urinary retention
- Aspiration pneumonia
- Chronic relapsing polyradiculoneuropathy and chronic unremitting polyradiculoneuropathy
- Relapse
Investigations
- CSF study: usually shows albumino-cytological dissociation. The protein concentration becomes elevated in 75% cases; this finding may not appear until 1-2 weeks after the onset of clinical manifestations. The protein may be as high as 400-500 mg/dl, although the cell count remains normal or only slightly raised.
- Motor nerve conduction velocity-reduced
- Electromyogram (EMG): shows evidence of acute denervation of muscle.
- Serum creatine phosphokinase level mildly elevated or normal.
- Sural nerve biopsy shows segmental demyelination, focal inflammation, and Wallerian degeneration, which is confirmatory.
- Stool culture serological test for campylobacter jejuni.
Treatment
Mainly supportive with physiotherapy to assist chest drainage and prevention of contractures (to prevent foot drop) with good supportive treatment; .90% of children will recover. Respiratory insufficiency secondary to paralysis of intercostal muscle is the most serious complication. Involvement of bulbar muscles and respiratory insufficiency may need tracheal intubation and assisted ventilation until recovery. Recovery has been reported to occur after .8 months of complete ventilatory support.
- specific treatment:
IVIG (haemoglobin): a total of 2g/kg to be given through IV infusion for 3-5 days a single daily dose.
Plasmapheresis - rehabilitation
Prognosis
Full recovery: approximately 85% (within 6-12 months) permanent neurological deficit: approximately 15%. The clinical course is benign, and spontaneous recovery begins within2-3 weeks. most patients regain full muscular strength, although some are left with residual or recur. Less than 5% of cases die either from autonomic involvement with cardiac arrhythmias, arterial pressure instability, respiratory insufficiency, or from the complication of assisted ventilation.