Systemic Lupus Erythematosus
The systemic lupus erythematosus (SLE, lupus is a rare disease with a prevalence that ranges from about 0.03% in Caucasians to 0.2% in Afro-Caribbeans. Some 90% of affected patients are female and the peak age at onset is between 20 and 30 years. SLE is associated with considerable morbidity and a fivefold increase in mortality compared to age- and gender-matched controls, mainly because of an increased risk of premature cardiovascular disease.
Pathophysiology
The cause of systemic lupus erythematosus is incompletely accepted but genetic factors play an important role. There is a higher concordance in monozygotic twins and the disease is strongly associated with polymorphic variants at the HLA locus. In a few instances. SLE is affiliated with inherited mutations in complement components C1q, C2 and C4, in the immunoglobulin receptor FcɤRlllb or in the DNA exonuclease TREX1.
Genome-wide association studies have identified common polymorphisms near several other genes that predispose to SLE, most of which are involved in regulating immune cell function. From an immunological standpoint, the characteristic feature of SLE is autoantibody production. These autoantibodies have specificity for a wide range of targets but many are directed against antigens present within the cell or within the nucleus.
This has led to the hypothesis that SLE may occur because of defects in apoptosis or in the clearance of apoptotic cells, which causes inappropriate exposure of intracellular antigens on the cell surface, leading to polyclonal B- and T-cell activation and autoantibody production. This is supported by the fact that environmental factors that cause flares of lupus, such as ultraviolet light and infections, increase oxidative stress and cause cell damage.
Whatever the underlying cause, autoantibody production and immune complex formation are thought to be important mechanisms of tissue damage in active SLE, leading to vasculitis and organ damage.
Clinical features
Symptoms such as fever, weight loss and mild lymphadenopathy may occur during flares of disease activity, whereas others such as fatigue and low-grade joint pains can be constant and not particularly associated with active inflammatory disease.
Arthritis
Arthralgia is a common symptom, occurring in 90% of patients, and is often associated with early morning stiffness. Tenosynovitis may also occur but clinically apparent synovitis with joint swelling is rare. Joint deformities may arise (Jaccoud’s arthropathy) as the result of tendon damage but joint erosions are not a feature.
Raynaud’s phenomenon
Raynaud s phenomenon (p. 504) is common and may antedate other symptoms by months or years. SLE can present with Raynaud’s phenomenon, along with arthralgia or arthritis. Secondary Raynaud’s phenomenon associated with SLE and other AICTDs needs to be differentiated from primary Raynaud’s phenomenon, which is common in the general population (up to 5%). Features in favour of secondary Raynaud’s phenomenon include age at onset of over 25 years, absence of a family history of Raynaud’s phenomenon, and occurrence in a male.
Examination of capillary nail-fold loops using an ophthalmoscope (and oil placed on the skin) can show loss of the normal loop pattern, with capillary fallout’ and dilatation and branching of 10ops: these features support either a diagnosis of systemic sclerosis or severe primary Raynaud’s phenomenon. If Raynaud s phenomenon is severe, digital ulceration can occur.
Skin
The skin is commonly involved in SLE, and many SLE skin eruptions are precipitated by exposure to ultraviolet light. The main types of skin involvement are:
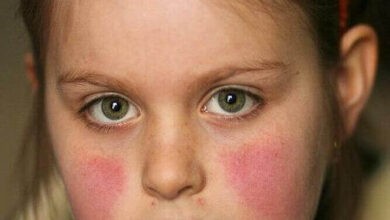
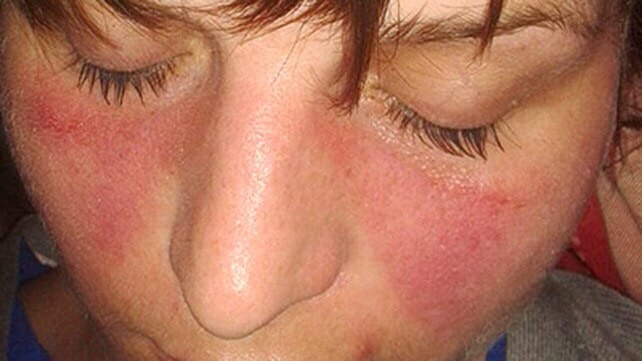
- The classic facial rash (up to 20% of patients). This is erythematous, raised and painful or itchy, and occurs over the cheeks with sparing of the nasolabial folds. Rosacea is a mimic of this rash.
- A discoid rash characterised by hyperkeratosis and follicular plugging, with scarring alopecia if it occurs on the scalp
- Diffuse, usually non-scarring alopecia, which may also occur with active disease.
- Urticarial eruptions.
- Livedo reticularis (Fig. 24.46), which is also a feature of antiphospholipid syndrome (p. 977) and can become frankly vasculitic, if severe.
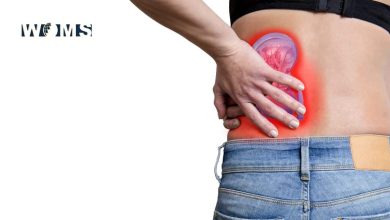
Κidney
Renal involvement is one of the main determinants of prognosis and regular monitoring of urinalysis and blood pressure is essential. The typical renal lesion is proliferative glomerulonephritis (p. 397), characterised by heavy haematuria, proteinuria and casts on urine microscopy.
Cardiovascular
The most common manifestation is pericarditis. Myocarditis and Libman-Sacks endocarditis can also occur. The endocarditis is due to accumulation on the heart valves of sterile fibrin-containing vegetations, which is thought to be a manifestation of hypercoagulability associated with antiphospholipid antibodies.
The risk of atherosclerosis is greatly increased, as is the risk of stroke and myocardial infarction. This is thought to be multifactorial due to the adverse effects of inflammation on the endothelium chronic glucocorticoid therapy and the procoagulant effects of antiphospholipid antibodies
Lung
Lung involvement is common and most frequently manifests as pleuritic pain (serositis) or pleural effusion. Other features include pneumonitis, atelectasis, reduced lung volume and pulmonary fibrosis that leads to breathlessness. The risk of thromboembolism is increased, especially in patients with antiphospholipid antibodies.
Neurological
Fatigue, headache and poor concentration are common and often occur in the absence of laboratory evidence of active disease. More categorical features of cerebral lupus include visual hallucinations, chorea, organic psychosis, transverse myelitis and lymphocytic meningitis.
Haematological
Neutropenia, lymphopenia, thrombocytopenia and haemolytic anaemia may occur, due to antibody-mediated destruction of peripheral blood cells. The degree of lymphopenia is a good guide to disease activity.
Gastrointestinal
Mouth ulcers may appear and may or may not be painful. Peritoneal serositis can cause acute pain. Mesenteric vasculitis is a serious complication. which can present with abdominal pain, bowel
infarction or perforation. Hepatitis is a recognised, though rare, feature.
Paediatric disease
Renal disease and cutaneous manifestations are more frequent in juvenile-onset SLE compared to disease in adults. Similarly, there is subsequently a higher incidence of renal disease, malar rash, Raynaud’s phenomenon, cutaneous vasculitis and neuropsychiatric manifestations than in adults.
Investigations
The diagnosis is based on a combination of clinical features and laboratory abnormalities. To fulfil the classification criteria for SLE at least 4 of the 11 factors shown in Box 24.63 must be present or have occurred in the past. Checking of ANAS, antibodies to ENAS and complement, routine haematology, biochemistry and urinalysis are mandatory.
Patients with active SLE test positive for ANA. Some authorities believe that ANA-negative SLE occurs (e.g. in the presence of antibodies to Ro) but others regard SLE as necessarily ANA-positive; the issue may be more to do with the sensitivity of the ANA assay at any given time in a disease course. Anti-dsDNA antibodies are positive in many, but not all, patients and are tested at the time of diagnosis by most laboratories using ELISA. ELISA has low specificity, whereas testing for anti-DSDNA antibodies using Crithidia luciliae is highly specific.
Patients with the active disease tend to have low levels of C3 due to complement consumption, but in some people, low C3 and C4 may be the result of inherited complement deficiency in C1, C2 or C4 that predisposes to SLE (p. 66). Studies of other family members can help to differentiate inherited deficiency from complement consumption. An increased ESR, leucopenia and lymphopenia are typical of active SLE, along with anaemia. Haemolytic anaemia and thrombocytopenia. CRP is often normal inactive SLE, except in the presence of serositis; thus an elevated CRP suggests infection.
Management
The therapeutic goals are to educate the patient about the nature of the illness, to control symptoms and to prevent organ damage and maintain normal function. Patients should be advised to avoid sun and ultravoilet light exposure and to employ sunblocks (sun protection factor 25-50).
Mild to moderate disease
Patients with mild disease restricted to skin and joints can sometimes be managed with analgesics, NSAIDs and hydroxychloroquine. Frequently, however, glucocorticoids are also necessary (prednisolone 5-20 mg/day), often in combination with Immunosuppressants such as methotrexate, azathioprine or mycophenolate mofetil (MMF). Increased doses of glucocorticoids may be required for flares inactivity or complications such as pleurisy or pericarditis. The monoclonal antibody belimumab which targets the β-cell growth factor BLyS has recently been shown to be effective in patients with active SLE who have responded inadequately to standard therapy.
Severe and life-threatening disease
High-dose glucocorticoids and immunosuppressants are required for the treatment of renal, CNS and cardiac involvement. A commonly used regimen is pulsed methylprednisolone (10 mg/kg IV) plus cyclophosphamide (15 mg/kg IV), repeated at 2-3-weekly intervals for six cycles. Cyclophosphamide may cause haemorrhagic cystitis but the risk can be minimised by good hydration and co-prescription of mesna (2-mercaptoethanol sulfonate), which binds its urotoxic metabolites. Because of the risk of azoospermia and premature menopause, sperm or oocyte collection and storage need to be considered prior to treatment with cyclophosphamide.
MME has been used successfully with high-dose glucocorticoids for renal involvement with results similar to those of pulsed cyclophosphamide but fewer adverse effects. Belimumab in combination with standard therapy significantly decreases disease activity in SLE patients and is safe and well-tolerated. Its role in patients with renal and neurological disease is still under investigation. Rituximab has been reported as being effective in selected cases, through randomised controlled trials have not shown significant overall efficacy
Maintenance therapy
Following control of acute disease, a typical maintenance regimen is an oral prednisolone in a dose of 40-60 mg daily, gradually reducing to 10-15 mg/day or less by 3 months. Azathioprine (2-2.5 mg/kg/day), methotrexate (10-25 mg/week) or MMF (2-3 g/day) should also be prescribed. The long-term aim is to continue the lowest dose of glucocorticoid and immunosuppressant to maintain remission. Cardiovascular risk factors, such as hypertension and hyperlipidaemia, should be controlled and patients should be advised to stop smoking. Patients with SLE and the antiphospholipid antibody syndrome, who have had the previous thrombosis, require life-long warfarin therapy. SLE patients are at risk of osteoporosis and hypovitaminosis D. and should be screened with biochemistry and DXA scanning accordingly.