Idiopathic Thrombocytopenic Purpura (ITP)
Idiopathic Thrombocytopenic Purpura (ITP) is the most common bleeding disorder of children where platelets are coated by a circulating antibody, developed against platelet glycoprotein antigens and eventually destroyed in the spleen.
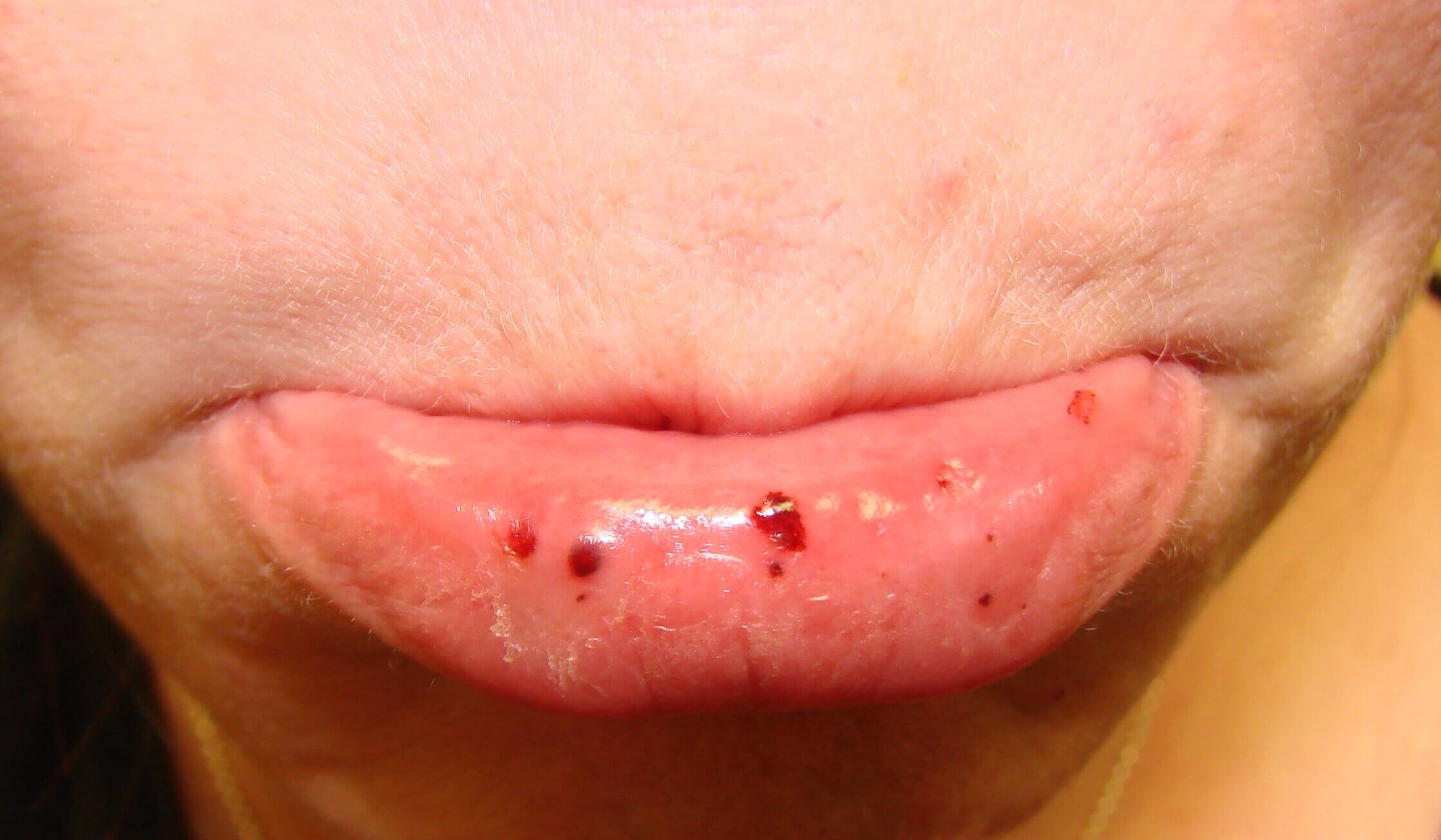
Purpura, or purple bruises on the skin or mucous membranes within the mouth, is common in people with ITP. These bruises might sometimes appear as petechiae, which are little red or purple dots on the skin. Petechiae might resemble a rash.
Both toddlers and adults can get ITP. There appear to be disparities in the development of ITP between men and women at different ages. ITP may be more common in women when they are younger. It may be more common in guys as they get older. After a general viral illness, children are more likely to develop this syndrome. ITP has also been connected to certain infections like chickenpox, mumps, and measles.
Types
- Acute Idiopathic Thrombocytopenic Purpura (ITP)
- Chronic Idiopathic Thrombocytopenic Purpura (ITP) (persistent thrombocytopenia >12 months)
Aetio-Pathogenesis
The aetiopathogenesis of Idiopathic Thrombocytopenic Purpura is unknown. However, in about 50-60% cases, this platelet destruction may follow an episode of viral upper respiratory tract infection about 2-3 weeks prior to the bleeding episodes.
The viruses commonly involved are Epstein Barr virus (EBV), Rubella, Varicella, Measles, Parvovirus B19 and Influenza. Antiplatelet antibodies are produced by the viruses, and these coat the circulating platelets, which are then killed and expelled by the spleen
Clinical manifestations
Sudden onset of mucocutaneous bleeding (distinguishable from coagulopathy) in a previously healthy child
- Skin: Petechiae, purpura, ecchymoses and easy bruising
- Mucosa: Bleeding from gum, nose, conjunctiva, menorrhagia
- Internal bleeding e.g. haematuria, haematemesis & melaena, haemoptysis and intracranial bleeding are uncommon but can occur in severe thrombocytopenia and may be fatal
Physical Examination
- Evidence of bleeding in
- Skin e.g. petechiae purpura or ecchymosis
- Mucosa e.g. nose. gum, oral cavity, conjunctiva
However, patients neither have significant pallor, hepatosplenomegaly, lymphadenopathy nor bony tenderness.
Differential Diagnosis
- Henoch Schonlein purpura
- Dengue haemorrhagic fever (DHF)
- Leukaemia
- Aplastic anaemia
- Meningococcal septicaemia
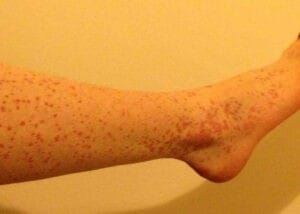
1. Skin rash of Henoch-Schonlein purpura
These rashes are characterized as palpable purpura that evolves from red to purple and then to rusty brown before they eventually fade. They are non-tender, do not blanch on pressure. Lesions usually involve buttock, lower extremities and the hands (waist down distribution).
2. Skin lesions of acute meningococcaemia
Lesions consist of tender pink macules. petechiae and purpura. These are most prominent on the extremities and may form areas of frank necrosis.
3. Skin lesions of Dengue fever
Transient macular rash in first 1-2 days, maculopapular. scarlet morbilliform, from day 3-5.
Acute Idiopathic Thrombocytopenic Purpura (ITP): Clinical severity
- Mild: Bruising and petechiae, occasional minor epistaxis, very little interference with daily living
- Moderate: More severe skin and mucosal lesions. Epistaxis and menorrhagia have become increasingly problematic.
- Severe: Bleeding episodes e.g. menorrhagia, epistaxis, melaena, requiring transfusion or hospitalization, Symptoms interfere seriously with the quality of life
Diagnosis
ITP is diagnosed by-
- Excluding other causes of thrombocytopenia.g. acute leukaemia, aplastic anaemia
- Analysing the clinical & laboratory data of the case
Investigations
- CBC&PBF
- Haemoglobin: Usually normal unless massive haemorrhage
- TC and DC of WBC: Usually normal
- Platelet count: Low
- PBF: RBC (normal), WBC (mature). No blast cells. Larger platelets may be seen
- Coagulation profile (usually not required):
- Bleeding time (BT): Prolonged
- Prothrombin time (PT): Normal
- aPTT: Normal
- Bone marrow study: Generally, not required for patients with isolated thrombocytopenia, who fit the diagnostic criteria above, but indicated, when-
- Patients fail to respond to the recommended therapy for ITP
- Cell lines, other than platelets are affected
- The steroid is planned to treat the case
- Bone marrow study findings: Megakaryocytes an increased. Erythroid and myeloid cellularity, as well as myeloid erythroid ratio, are normal
Differences between acute ITP leukaemia and aplastic anaemia
A. Clinical
| Parameters | Acute ITP | Acute leukaemia | Aplastic anaemia |
| Skin/gum/nose bleeding | Present | Present | Present |
| Fever | Usually absent | Usually present | Usually present |
| Appearance | Normal | Sick, irritable | Sick |
| Pallor | Insignificant | Severe | Severe |
| Lymphadenopathy | Absent | Present | Absent |
| Bony tenderness | Absent | Present | Absent |
| Liver size | Normal | Enlarged | Normal |
| Spleen size | Normal | Enlarged | Normal |
B. Laboratory
| Parameters | Acute ITP | Acute leukaemia | Aplastic anaemia |
| Haemoglobin | Normal or slightly reduced | Low | Low |
| WBC count | Normal | Usually very high |
Low |
| Platelet counts | Low | Low | Low |
| Blood film | Normal | Presence of Blast cells | Pancytopenia but cell morphology are normal |
| Bone marrow findings | Increased megakaryocytes | Нуper cellular Blast cells of>25% is diagnostic | Markedly hypocellular marrow, devoid of haematopoietic cells |
Treatment
Acute ITP is a self-limiting disease and > 80% of children require no therapy. Only-
- Counselling to parents & patients about the disease
- Close observation of the patients
- Avoidance of aspirin, NSAID and
- Restriction from physical contact activities is recommended.
However, when the platelet count falls <20,000/cmm, treatment is required to induce a rapid rise of platelets to the safe level. The following drugs are recommended in this situation-
- IVIG: The treatment of choice for severe, acute bleeding. Dose: 0.8-1.0 g/kg/day for 1-2 days. It induces a rapid rise of platelet count (>20 x 109/L) in 95% of patients within 48 hours. IVIG blocks the Fe-receptors in macrophages and thereby prevent phagocytosis of antibody-coated platelets
- Prednisolone: Patients with clinically significant but non-life-threatening bleeding may benefit from this
Dose: 1-4 mg/kg/day, continued for 2-3 weeks until platelet count is > 20 x 109/L - IV anti-D to Rh-positive patients: Single dose: 50-75 μg/kg, induces a rapid rise of platelet count >20 x 109/L within 48-72 hours in 80-90% of patients. This
the antibody binds to the D antigen on RBCs which then interferes with the removal of antibody-coated platelets in spleen, resulting in improvement in thrombocytopenia - Other options-
- Splenectomy: Reserved for refractory
thrombocytopenia with life-threatening haemorrhage - Platelet transfusion: Usually not indicated in acute ITP, as the transfused platelets will be liable to be coated by the antiplatelet-antibodies and destroyed If it is life-saving, transfuse along with IVIG or steroid
- Splenectomy: Reserved for refractory
Prognosis
About 80% of children with acute ITP will achieve remission and 20% may turn into chronic ITP. The predictors of chronicity are-
- Female gender
- Age > 10 years at presentation
- Insidious onset of bruising
- Presence of other autoantibodies
Summary
The majority of patients with ITP do not have a significant or life-threatening condition. Without therapy, acute ITP in children usually disappears in six months or less. Chronic ITP can persist a long time.
Even those who have severe instances of the disease can live for decades. Many persons with ITP can safely manage their illness without long-term consequences or a shorter life expectancy.
FAQs:
1.Is ITP dangerous to life?
In many cases, it is not dangerous or life-threatening. In case of acute ITP seen in children, it will heal itself within six months or less without any treatment while chronic ITP can last for many years.
2.Can stress cause ITP?
Some research says that psychical or psychological stress and resultant oxidative stress in our bodies can trigger ITP in children. Similarly, exacerbate fatigue and platelet disorder for a long time can also cause ITP.
3.Is ITP hereditary?
ITP is not considered to be a hereditary disease. If the family members have ITP then their blood sample must be verified by the hematologist that it is due to an autoimmune cause but not due to a hereditary cause.
4.How to diagnose ITP?
ITP is diagnosed by the low count of platelet in the complete blood count. For further investigation bone marrow biopsy is also done.